What is it?
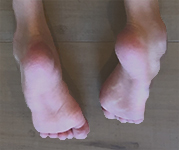 HSAN1, previously known as HSN1, is a rare inherited peripheral neuropathy. Each child of an affected parent has a 50 percent chance of inheriting the affected gene. HSAN1 patients develop severe loss of sensation to temperature, pressure, and pain. The loss starts in the extremities, usually beginning in the feet. This sensory loss often leads to painless injuries, chronic skin ulcers, bone destruction, bone infections, pain, amputation of fingers and toes, and, eventually, difficulties with movement.
HSAN1, previously known as HSN1, is a rare inherited peripheral neuropathy. Each child of an affected parent has a 50 percent chance of inheriting the affected gene. HSAN1 patients develop severe loss of sensation to temperature, pressure, and pain. The loss starts in the extremities, usually beginning in the feet. This sensory loss often leads to painless injuries, chronic skin ulcers, bone destruction, bone infections, pain, amputation of fingers and toes, and, eventually, difficulties with movement.
HSAN1 symptoms typically begin in the late teens. The diagnosis is made by genetic testing. HSAN1 is progressively debilitating and severely disabling, and should be treated urgently. Many affected patients often notice weakness and muscle atrophy, and require surgeries or walking aids such as canes, braces, or wheelchairs.

For more on HSAN1, view slides provided by the Deater Foundation, Inc.
HSAN1 can be treated
 HSAN1 is caused by mutations in the genes SPTLC1 and SPTLC2. These genes encode the serine palmitoyltransferase (SPT) enzyme. SPT controls the production of sphingolipids, a fat important for normal neuron function. In HSAN1, SPTLC mutations alter SPT so that the enzyme produces abnormal fats called deoxysphingoid bases (DSBs). These accumulate in sensory neurons, impairing the cells’ proper function and leading to HSAN1 symptoms.
HSAN1 is caused by mutations in the genes SPTLC1 and SPTLC2. These genes encode the serine palmitoyltransferase (SPT) enzyme. SPT controls the production of sphingolipids, a fat important for normal neuron function. In HSAN1, SPTLC mutations alter SPT so that the enzyme produces abnormal fats called deoxysphingoid bases (DSBs). These accumulate in sensory neurons, impairing the cells’ proper function and leading to HSAN1 symptoms.
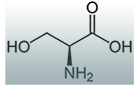
A targeted treatment for HSAN1 has been discovered. A normal dietary amino acid, L-serine, when taken in extra-large doses improved symptoms and nerve function in adults with HSAN1. In a clinical trial, L-serine supplementation clearly reduced levels of the toxic DSBs in patients. L-serine helped with sensory symptoms, and appeared to increase arm and leg strength in people who took it.
 Going forward, researchers and advocates are working to create an HSAN1 patient registry. This resource would collect information and biospecimens such as blood and DNA from people with the disease. Such a registry would help patients and doctors with fact-based guidance about treatment. It would aid researchers to understand the disease and potential treatments. Right now, people with confirmed HSAN1 are encouraged to participate in the Mass General Hospital’s Neuropathy Gene Registry, which is available to patients with all types of genetic neuropathies.
Going forward, researchers and advocates are working to create an HSAN1 patient registry. This resource would collect information and biospecimens such as blood and DNA from people with the disease. Such a registry would help patients and doctors with fact-based guidance about treatment. It would aid researchers to understand the disease and potential treatments. Right now, people with confirmed HSAN1 are encouraged to participate in the Mass General Hospital’s Neuropathy Gene Registry, which is available to patients with all types of genetic neuropathies.
Also in the works is a study of L-serine supplementation in children. Even though HSAN1 symptoms do not appear until the teen years or later, the harm from DSBs likely begins in childhood. The goal of the study will be to see if taking L-serine in childhood or early adulthood prevents or slows the appearance of symptoms later.
HSAN1 is very rare, and frequently goes undiagnosed, making it difficult to determine how many people suffer from this form of neuropathy. One estimate suggests it affects only a few hundred people worldwide. However, harmful DSBs have been detected in other, more common forms of small-fiber neuropathy, including diabetic sensory neuropathy and chemotherapy-induced peripheral neuropathy. That raises the possibility that these diseases might also respond to L-serine supplementation.
A patient’s story
From Alexis E., age 18, whose father and grandmother have HSAN1:
Learn more about HSAN1. Slides provided by the Deater Foundation, Inc.
More resources
Deater Foundation, Inc., is the leading funder of HSAN1 research, including the clinical trials of L-serine.
On Facebook: Deater Foundation, Inc.; Neuropathy Talk: HSAN Type 1
Key publications
Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1.
V Fridman, S Suriyanarayanan, P Novak, W David, EA Macklin, D McKenna-Yasek, K Walsh, R Aziz-Bose, AL Oaklander, R Brown, T Hornemann, F Eichler.
Neurology. 2019 Jan 22;92(4):e359-e370.
Natural history and biomarkers in hereditary sensory neuropathy type 1.
V Fridman, AL Oaklander, WS David, EA Johnson, J Pan, P Novak, RH Brown Jr, FS Eichler.
Muscle Nerve. 2015 Apr;51(4):489-95.
Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1.
K Garofalo, A Penno, BP Schmidt, H-J Lee, MP Frosch, A von Eckardstein, RH Brown, T Hornemann, FS Eichler.
J Clin Invest. 2011 Dec;121(12):4735-45.